Introduction
MetalS2 is a software tool for the structural alignment of Minimal Functional Sites (MFSs) in metal-binding biological macromolecules.
Focusing on MFSs allows functional linkages between different proteins of known structure to be made with greater confidence than with metal-binding sites.
This is because the ligand neighbors play a crucial role in tuning the properties of the metal-binding site and, in particular, the reactivity of the metal ion.
A crucial feature of such a tool must be that it takes explicitly into account the fact that MFSs are built around metal sites.
Consequently the structural comparison should start from there with superimposition of metals.
MetalS2 continues its procedure to determine the best alignment of two MFSs with the superposition of the metal ions
(or of the geometric center of polymetallic cofactors) and the comparison of the position of donor atoms.
Consequently, the metal sites are always at the center of the structural alignment.
This intrinsically reflects the philosophy underlying the construction of MFSs.
MetalS2 supports the comparison of MFSs harboring different metals and/or with different nuclearity,
and is available both as a stand-alone programDownload MetalS² stand-alone program
and a web toolGo to MetalS² home.
Software Requirements
The MetalS2 web service requires Java Run-time Environment (JRE) installed on your local machine.
You can download the latest Java at java.com.
The service runs on most operating systems as well as on all common browsers.
If you experience any trouble using MetalS2, please feel free to contact us.
Getting Srarted
To align two sites, please, complete the following simple steps.
-
Specify the input structures
The first form allows you to specify two input structures that contain MFSs.
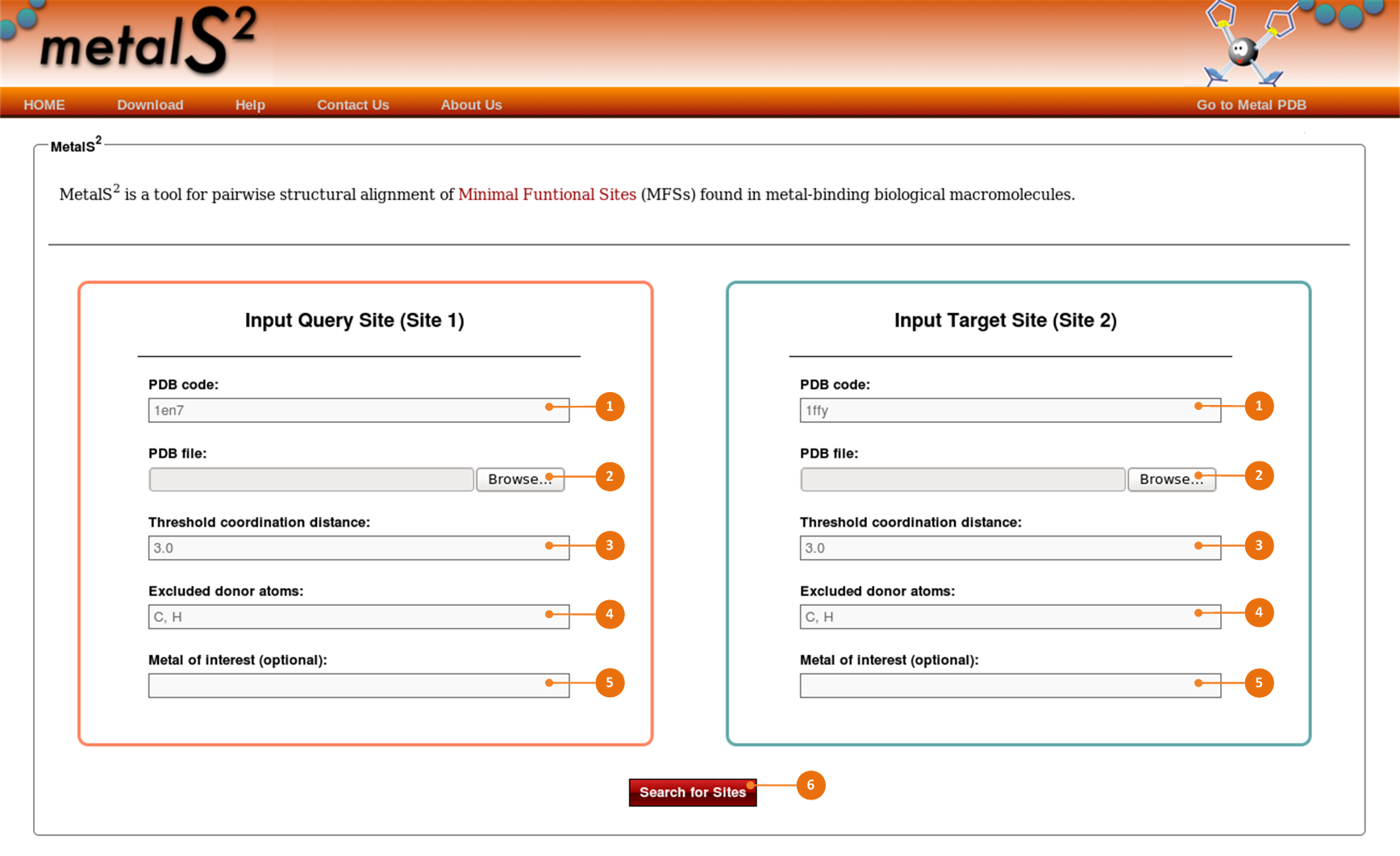
The MetalS2 portal can automatically search structures deposited in the PDB for MFSs, taking as input the corresponding PDB code.
To specify PDB structures, enter four-letter PDB code for each structure into the PDB code fields (1) in the left and right areas.
The PDB-codes are required to start the alignment procedure, except if you upload the structures as atomic coordinate files.
If structures are not available publicly, you can browse your local hard drive using the “Browse…” buttons (2) in the left and right areas
and upload the PDB-files of structures containing metal sites you want to align.
Use the next fields to adjust the search options. Changing the options may affect results of the procedure designed to extract metal sites from structures:
-
Change the default value of 2.8 Å in the “Threshold coordination distance” fields (3).
The ligands to each metal atom in each structure are identified, as having at least one non-hydrogen atom at a distance smaller than 2.8 Å from the metal.
-
Adjust the default list of atoms in the “Excluded donor atoms” fields (4) entering the chemical symbol(s) of the atom(s) (separated by commas).
“Excluded donor atoms” is a list of atoms that will not be identified as atoms that coordinate the metals, thus, will not be considered as donor atoms.
Defaults are C and H.
-
Enter the chemical symbol(s) of a specific metal(s) into the “Metal of interest (optional)” fields (5).
The search of metal sites will be only done near atoms of this type.
Click the “Search for Sites” button(6), and MetalS2 will automatically identify all MFSs present in the input structures.
-
Select sites
As soon as the input structures are processed you see all detected MFSs listed.
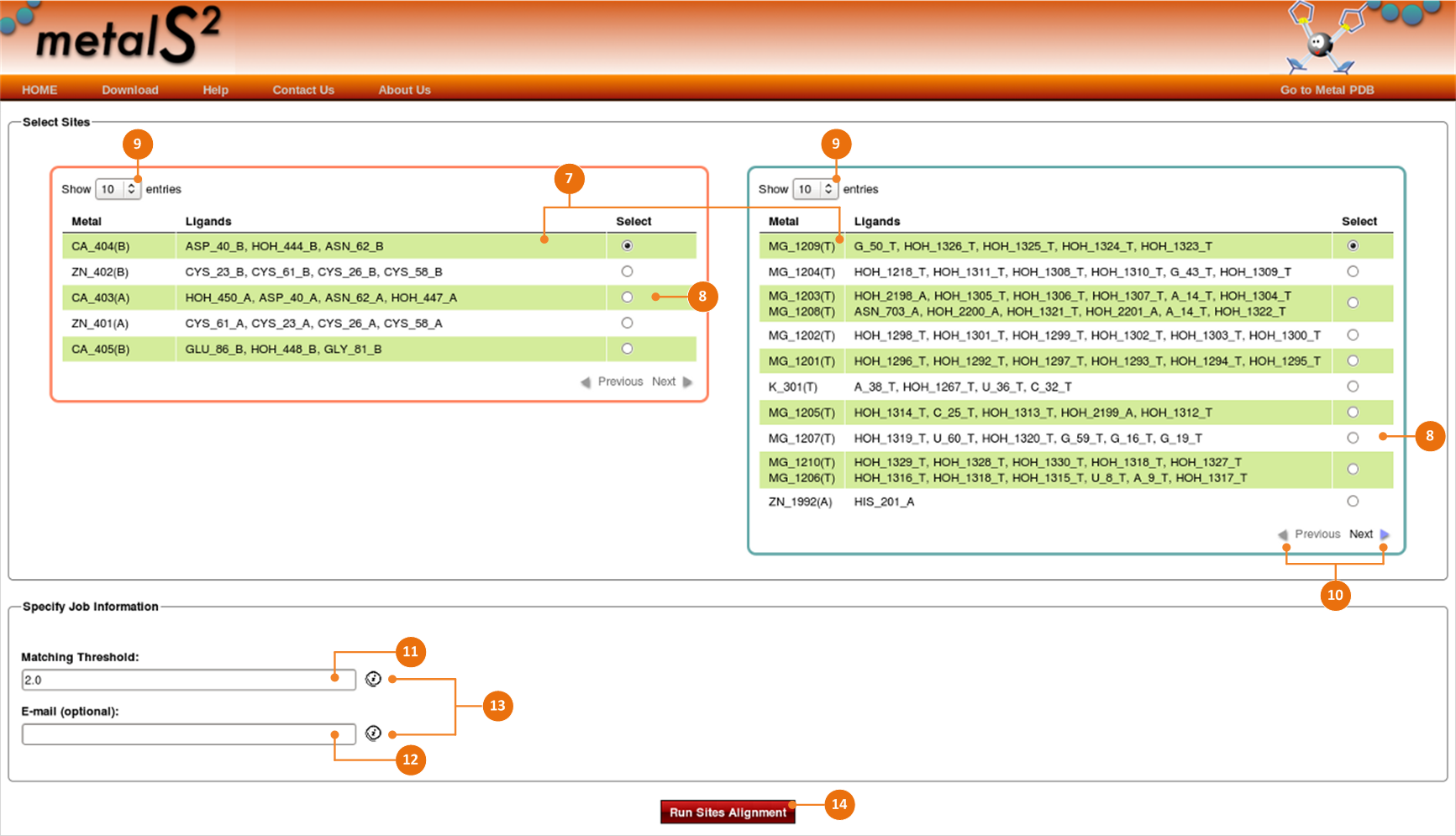
Sites are represented by records in the fields (7) each contains the information about the metal site:
- Metal section displays the metal(s) that belongs to a metal site. The metal is identified with its element type,
residue sequence number, and the name of chain to witch it binds.
- Ligands section displays the residues which bind the metal.
Each residue is identified with a residue name, residue sequence number, and a chain name.
Select two sites for which you want to perform alignment by checking the corresponding radio buttons in the “Select” column (8) in the left and right areas.
Only one site can be selected in each area.
By default, you see 10 search results per page. This number gives you the fastest search, but you can increase the number.
Hit the Show entries box (9) and choose the number of sites you would like to display.
You can see other pages using the Next/Previous navigation links (10).
When alignding atoms MetalS2 looks for the atom from the target site closest to the atom from query site.
We restrict the search within a radius of 2 Å around the atom of the query site by default.
You can change the default value in the “RMSD Threshold” field (11).
If there is no atom of the target structure in this range, the atom of the query structure will remain unmatched.
If you provide an email address (12), MetalS2 will send you a link to results when your job is finished.
There are tips (13) that help you get quickly extra information. You can see them tracking along with the cursor movement over the "info" icon.
Use the “Run Sites Alignment” button (14) to start the alignment procedure.
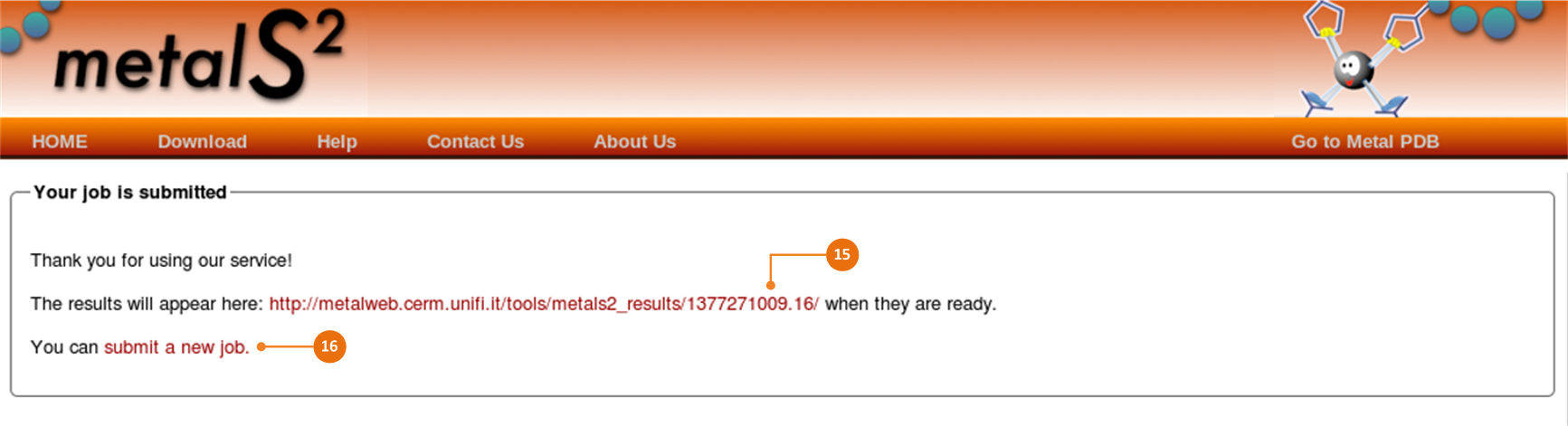
If the alignment is started successfully, you will see the page as shown. From this page you can access results following a provided link (15).
Note: Please, keep the link available if an email address has not been specified.
Review results
Results are shown in tree sections:
Scores and RMSD,
Structural alignment, and
Sequence alignment based on structural alignment.
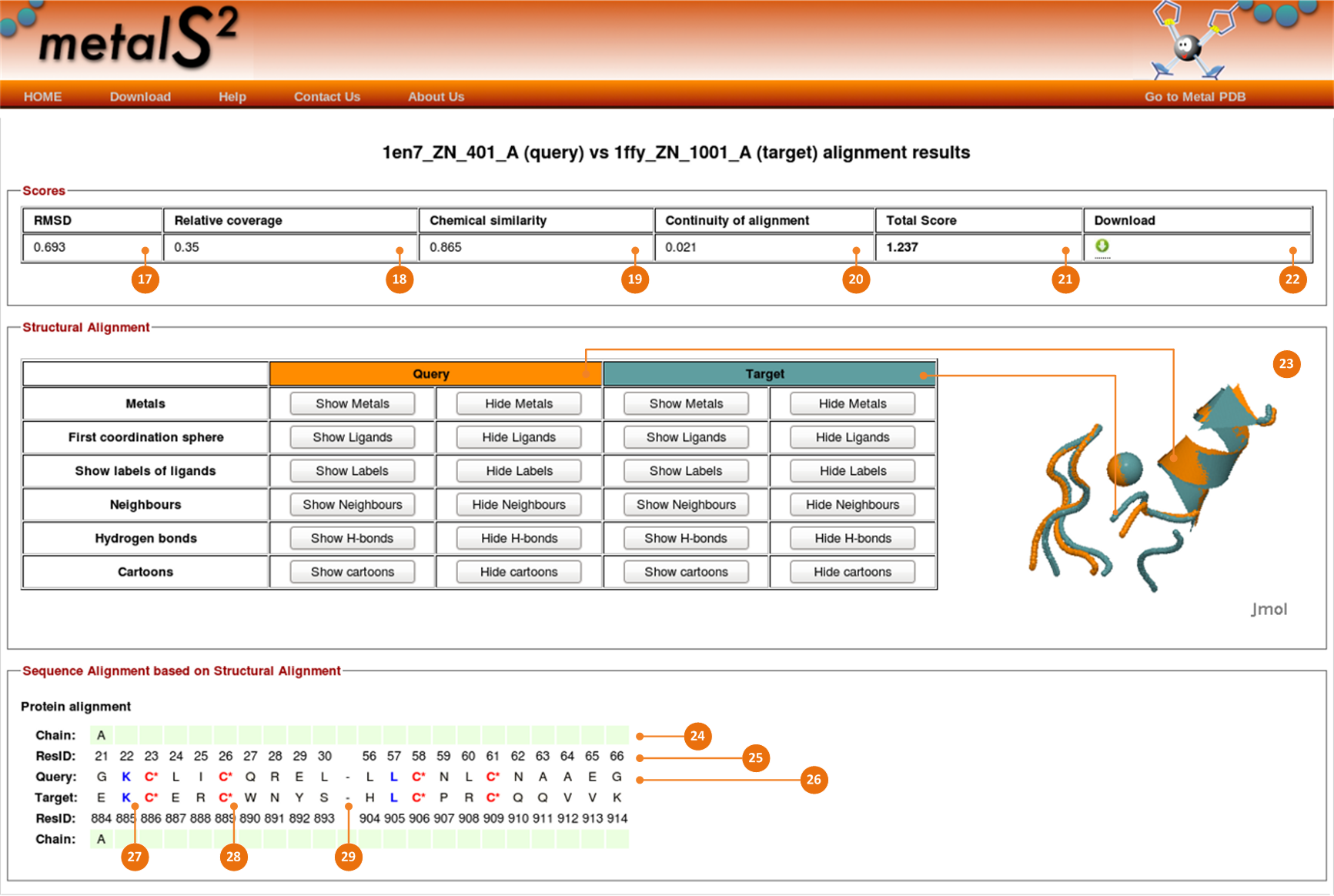
Scores and RMSD
The MetalS2 total score (21) is given as a summarized value of three contributions:
Relative coverage term (18), Sequence similarity term (19), and Fragmentation term (20).
Each term describes quantitatively an essential property of the structural alignment.
- A relative coverage term, depending on the ratio between the number of atoms put in correspondence and
the maximum possible number of atom correspondences for the sites being compared.
- A sequence similarity term, depending on the ratio between the similarity score computed using the BLOSUM62 matrix
for the sequence alignment derived from the Cα correspondences and the similarity score that would be obtained
if the two sites being aligned had identical sequences. To compute the last, we consider the sequence giving the lowest similarity score to itself.
For nucleic acids we used a simple scoring system that consists of a "reward" for a match (+5) and a "penalty" for a mismatch (-4).
- A fragmentation term, which takes into account how many fragments the alignment is broken into and how long each segment is.
The lower is the total score, the more the degree of similarity between structures.
A broad maximum at 2.75 can be taken as the threshold below which the MetalS2 total score indicates a good structural alignment.
Besides the scores MetalS2 provides the value of root mean square deviation (17) computed over the subset of aligned atoms.
You can also download an archive with resulting output files (see “MetalS2 output” for more details) by clicking on the
 button in the “Download” column (22).
The results.tgz archive will be downloaded to your local machine.
button in the “Download” column (22).
The results.tgz archive will be downloaded to your local machine.
Structural alignment
The structural alignment is shown as superposed 3D structures displayed using Jmol script. Note: Java is required to run the Jmol plugin.
The script highlights the structures in different colors to visually distinguish query and target sites (23).
The elements of query and target structures can selectively be shown and hidden to facilitate the analysis.
Furthermore, the Jmol applet is capable of enlarging and scaling down the view
to examine the structures and their superposition in more details.
Full documentation on the Jmol viewer can be found on the Jmol Wiki pages.
Sequence alignment based on structural alignment
Besides the information about structural similarities, MetalS2 provides information about sequence alignment based on structural alignment.
There is the aligned fragment of a query structure’s sequence in the upper part.
The aligned residues of a target sequence are placed below. The aligned sequence of a query structure preserves the sequence order, i.e.,
it has the same order as input sequences. The target sequence is in aligned order.
A sequence record consists of a three-line description.
The first line of the description gives a reference to a chain (24) which aligned residues belong to.
The second line is intended for sequence numbering (25).
The third line of the description shows the sequence of one-letter residue codes (26).
Equal residues (27) and equal ligands (28) are highlighted with blue and
red colours correspondingly. Interruptins of the consequent alignment is indicated by - (29).
The descriptions for query and target sequences are mirrored. Order of escription lines for the target sequence should be sought as reversed.
MetalS2 output
MetalS2 generates the downloadable archive with the output results. The content is organized as follows:
site1_vs_site2/
query_site
target_site
visualization.pml
score.txt
sequence.txt
-
The site1_vs_site2 directory holds the PDB-like formatted files with fitted coordinates of superimposed structures.
You can then feed the output files to a viewer to illustrate structures.
-
The score.txt text file contains a score value of alignment calculated using a scoring function.
-
The text file called sequence.txt contains a structure-based sequence alignment written to a text file.
Note: only residues that are structurally aligned are presented in the output sequences.
Usage of the stand-alone version
MetalS2 is also available as a stand-alone version which has all the functionality of the web version except the possibility to search for the input structures in the database.
The stand-alone version allows working with files stored on your local machine.
The tool produces the same output files described above (see MetalS2 output), which are placed in subdirectories each called by the combination of
names of the metal sites that were aligned (e.g., 1en7_vs_1ffy).
Synopsis
To start the process of alignment using a command-line interface, issue a command in the following form:
$python metalS2.py option filename option filename [--qm number] [--tm number] [-d distance] [output directory]
Options
It is mandatory to specify the input structures to start the alignment process.
The following options allow you to specify what the types of input files are in order to invoke the correct procedure of the inputs processing.
--qp Specify that the following file is a PDB that contains a query metal site.
--tp Specify that the following file is a PDB that contains a target metal site.
--qs Specify that the following file encloses a query metal-binding site.
--ts Specify that the following file encloses a target metal-binding site.
If your input is a PDB file with a number of sites and you know exactly which metal atom should enter into the composition of a metal site,
you can explicitly specify this metal using the following options:
--qm Specify a metal-binding site of interest in a query structure.
--tm Specify a metal-binding site of interest in a target structure.
The option is to be mandatory followed by a number and may be set for both PDB files.
The number is a residue sequence number of metal atom trapping metal-binding site structure. Default is all metals.
You can adjust your alignment procedure by setting the following options:
-d Set a distance cutoff value, in Angstroms, that allows controlling the upper bound of the area where two atoms may be considered as aligned.
The cutoff value can be any non-negative floating point number. The default value is 2.0 Å. A value of 0 prevents the program from running at all.
You can declare the location where you want to store the results of alignment. Add the relative path to the output directory to a command.
/relative/path/to/the/output/directory
If the output directory is omitted the results will be stored in a current directory.
The following options will give you a summary of the usage and available options. If you forget all the other options, remember these.
-h --help Print a brief reminder of command line usage and all available options.
-u --usage Print a usage summary.